Table 3.1 lists functional groups. Useful as a reference - no need to memorize it.
Classifications of hydrocarbons:
saturated: contains single bonds only (no pi\pi bonds)
unsaturated: contains pi\pi bonds
cyclic: contains one or more rings
acyclic: contains no ring
aromatic: contains a benzene ring or analogous structure
Cc1ccc(C(C)C)cc1
aliphatic: not aromatic (can be saturated or unsaturated)
IUPAC classification
alkanes: saturated. If acyclic, its formula is C_(n)H_(2n+2)\mathrm{C}_{n} \mathrm{H}_{2 n+2}
CC1=CCC(=C(C)C)C=C1
alkenes: contains >= 1\geq 1 double bond
alkynes: contains >= 1\geq 1 triple bond
Isomers: molecules with the same empirical formula but different structure.
Constitutional isomers differ in their bond connectivity.
CCCCC
Linear
CCC(C)C
Branched
CC(C)(C)C
CC(C)(C)C
IUPAC nomenclature. (In Table 3.3, memorize names for C_(1)-C_(13)\mathrm{C}_{1}-\mathrm{C}_{13} )
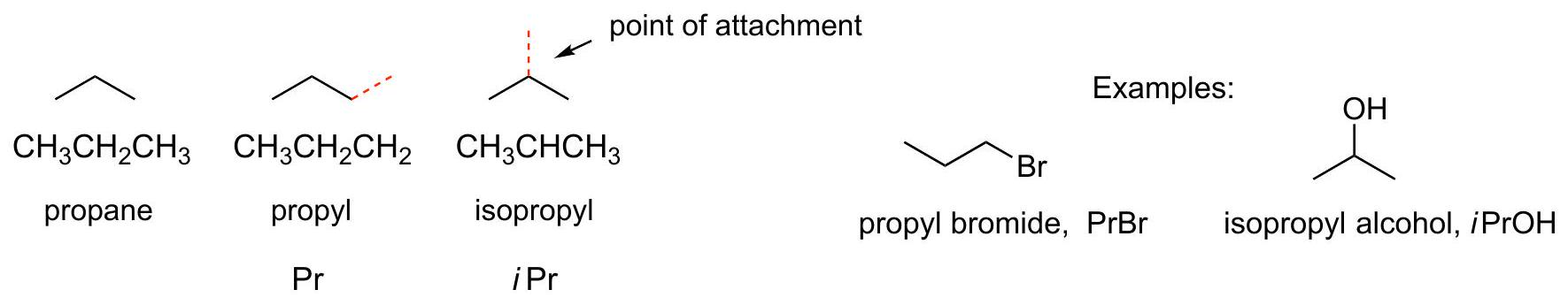
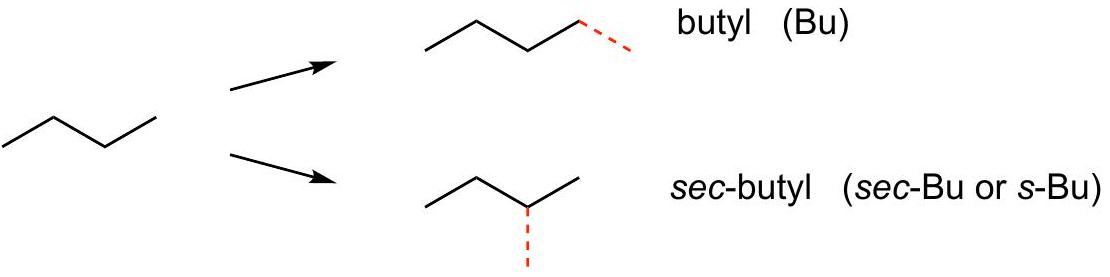
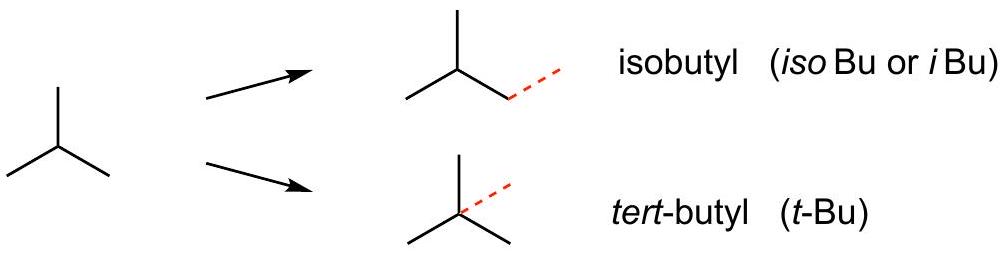
Simple alkyl groups: C_(3)H_(7)\mathrm{C}_{3} \mathrm{H}_{7} and C_(4)H_(9)\mathrm{C}_{4} \mathrm{H}_{9}
Two isomers of C_(4)H_(10)\mathrm{C}_{4} \mathrm{H}_{10} :
CCCCC
CC(C)C
C_(4)H_(9)\mathrm{C}_{4} \mathrm{H}_{9} groups derived from each C_(4)H_(10)\mathrm{C}_{4} \mathrm{H}_{10} isomer:
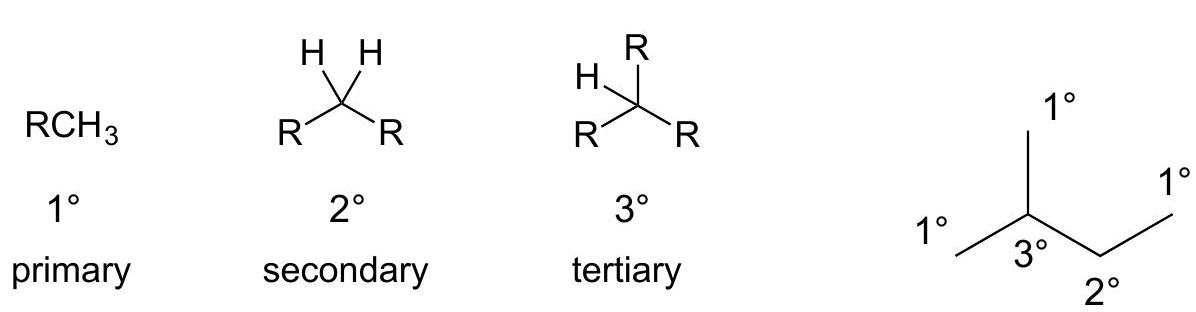
Degree of alkyl substitution on C ( RR is any alkyl group) 1^(@),2^(@),3^(@)1^{\circ}, 2^{\circ}, 3^{\circ} can be applied to C atoms or H atoms
a primary alkyl chloride
IUPAC names
CCCC(C)CCC(C)CC
Find the longest chain and name it.
Number the atoms in the chain, beginning at the end nearest a branch point.
Name each substituent & give it a number.
Write the name as a single word, with substituents in alphabetical order.
CCCC(C)C(C)CCC
4,7-dimethylnonane
CCCC(C)CCC(C)CC
3,6-dimethylnonane
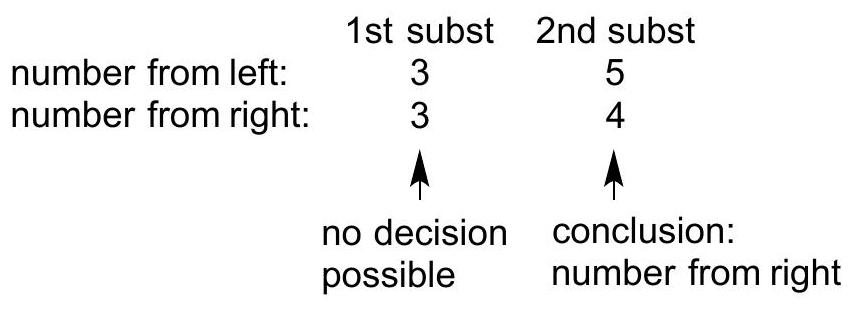
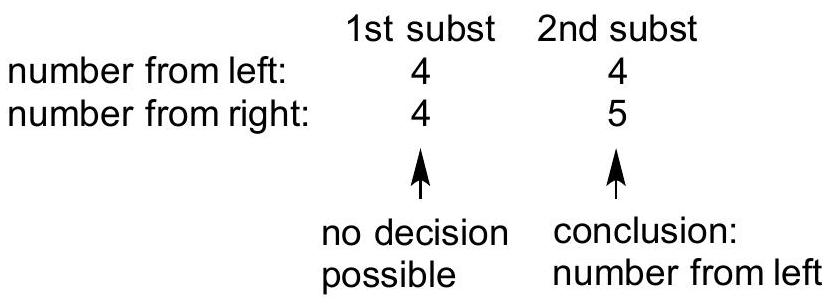
WRONG because the 1st substituent does not have the lowest possible number
CORRECT because the 1 st substituent does have the lowest possible number
CCC(CCCC(C)C)C[C@@H](Cl)CC
8-chloro-6-ethyl-2-methyldecane
In numbering the chain: if there are multiple substituents, number the chain in such a way that the first substituent has the lowest number. If a decision is not possible, then number the chain to minimize the number on the second subsituent, and so on, until a decision is reached.
Only as a last resort, number according to alphabetical order
CC(F)CC(C)CC(C)Cl
2-chloro-6-fluoro-4-methylheptane (based on the alphabet)
CC(Cl)CCC(C)C(C)F
6-chloro-2-fluoro-3-methylheptane (based on position of 2nd sub.)
CCCCC(C(CC)CCC)C(C)(C)CC
5-(1,1-dimethylpropyl)-4-ethylnonane
Name a branched substituent based on its longest chain, counting from the point of attachment to the main chain as carbon 1. Place the substituent name in parentheses. Use the first letter within the parentheses for alphebatizing, even if it starts with di, tri, etc.
IUPAC allows common names for simple branched substituents (isopropyl, tert-butyl, etc.).
CCCCC(CCC)C(C)C
4-isopropylheptane or 4-(1-methylethyl)heptane
Prefixes that are ignored in alphabetizing:
di, tri, tetra, ...
sec-, tert-
(exception: branched substituents - see example above)
Prefixes that are included in alphabetizing: iso, neo, cyclo
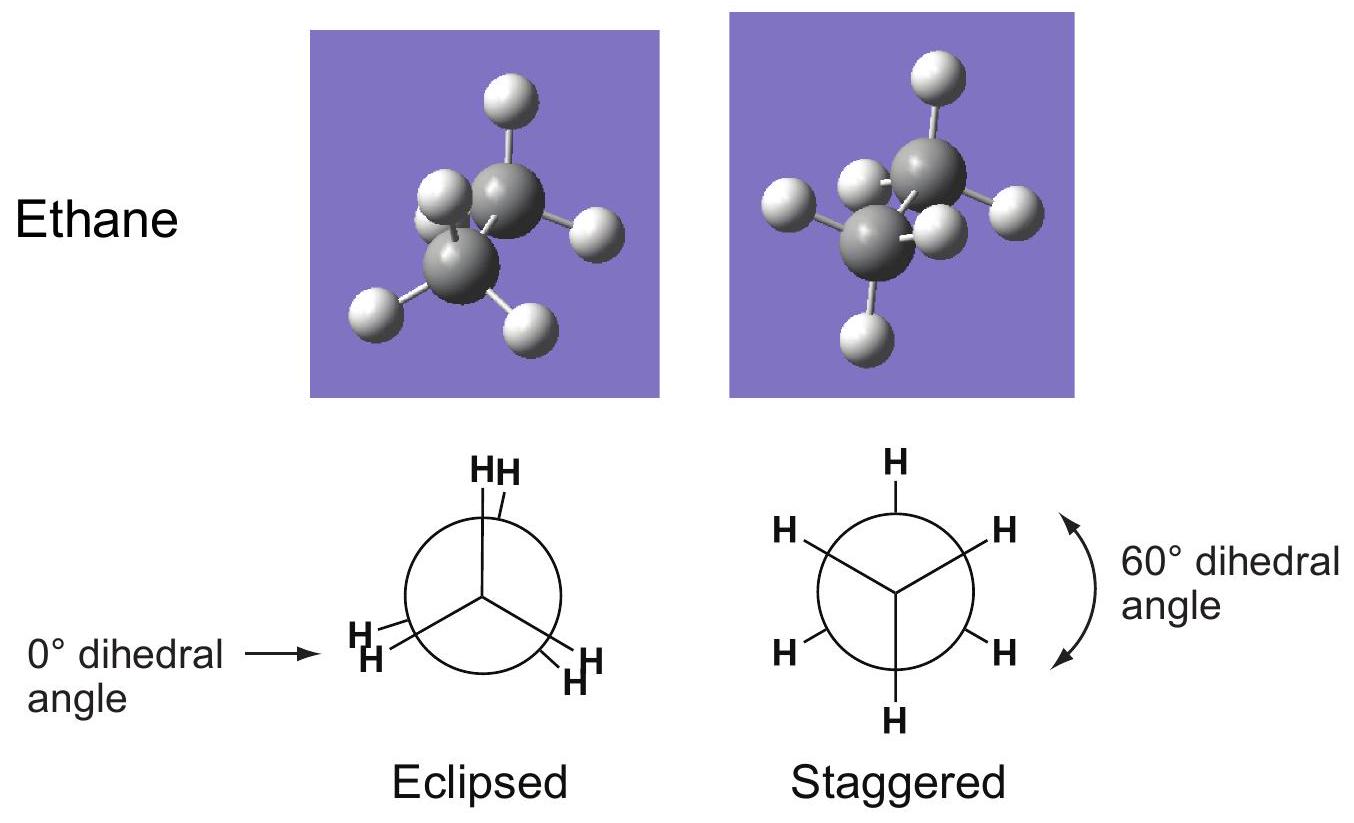
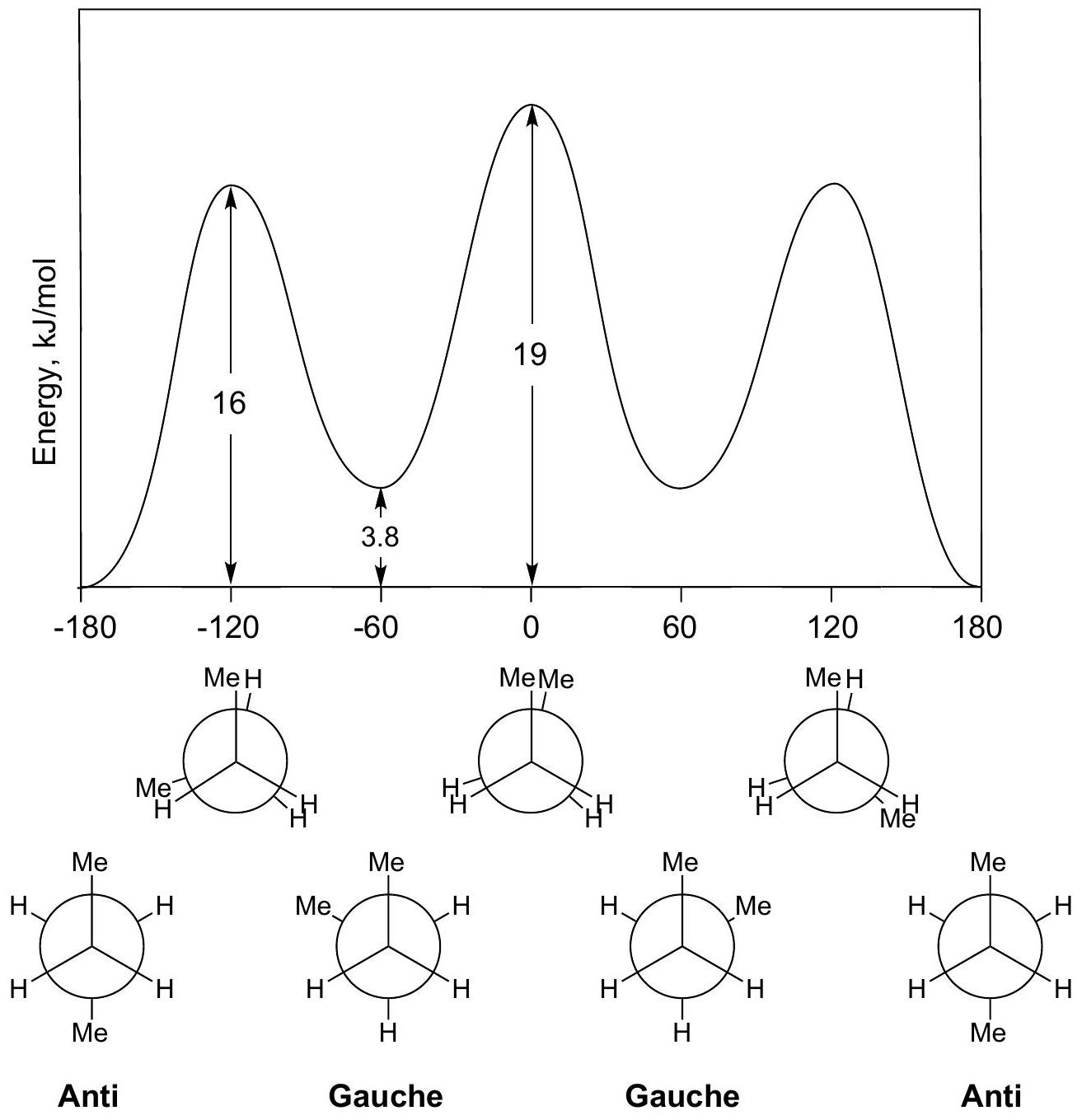
CONFORMATIONS OF ALKANES
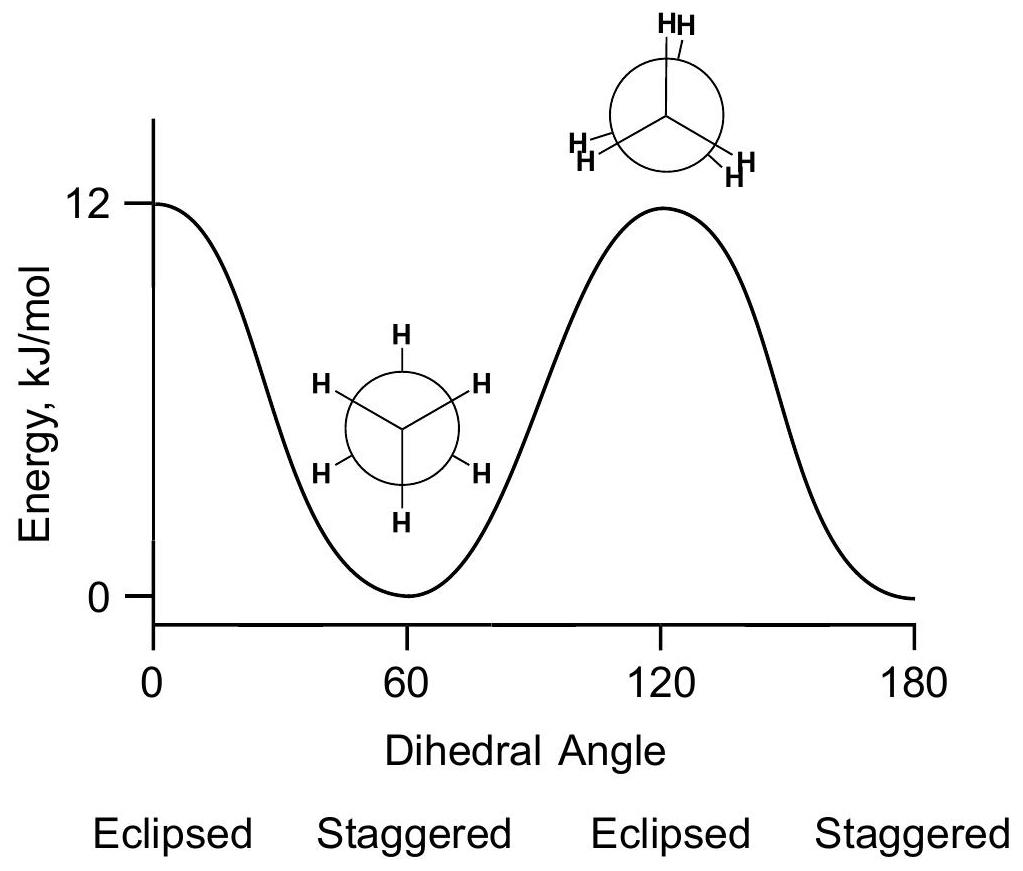
Useful approximation: the 12kJ//mol12 \mathrm{~kJ} / \mathrm{mol} barrier is considered to be the sum of 3H-H3 \mathrm{H}-\mathrm{H} eclipsing interactions. Then one H-H\mathrm{H}-\mathrm{H} eclipsing interaction contributes 4kJ//mol4 \mathrm{~kJ} / \mathrm{mol} to the barrier. The approximation is that we assume an H-H\mathrm{H}-\mathrm{H} eclipsing interaction contributes 4kJ//mol4 \mathrm{~kJ} / \mathrm{mol} in any molecule.
Why is the staggered geometry lower in energy than eclipsed?
Two models:
Steric interaction model: as two groups approach each other and their electron densities begin to overlap, electron-electron repulsion sharply raises the energy. The larger the group, the greater the steric repulsion.
Hyperconjugation model (called "torsional strain" by McMurry): the staggered geometry allows greater electron delocalization than the eclipsed, via interaction of filled with empty sigma\sigma orbitals.
For ethane, the concensus is that both effects are important. For molecules with interacting groups larger than H (Me, Et, iPr, tt-Bu, etc.), steric interactions are dominant.
Hyperconjugation, or delocalization of sigma\sigma orbitals, occurs throughout organic chemistry and is discussed briefly below. The description below is not in McMurry and will not appear on any exam or quiz.
As ethane rotates, an "electron delocalization window" alternately opens (staggered) and closes (eclipsed). In the staggered geometry, bonding and antibonding sigmaC-H\sigma \mathrm{C}-\mathrm{H} orbitals ( sigma_(CH)\sigma_{\mathrm{CH}} and sigma_(CH)\sigma_{\mathrm{CH}} ) on adjacent carbons interact to give a partial pi\pi bond between C atoms. Compared to a sigma_(CH)\sigma_{\mathrm{CH}} orbital, this gives a larger space in which electrons can move, encompassing 2 C atoms instead of one. By the Uncertainty Principle, delocalization lowers the energy of staggered ethane. This is not available to eclipsed ethane because of weak net overlap of sigma_(CH)\sigma_{\mathrm{CH}} and sigma^(**)_(CH)\sigma{ }^{*}{ }_{\mathrm{CH}} orbitals of adjacent eclipsed C-H bonds.
Electrons are not delocalized. Energy is not lowered.
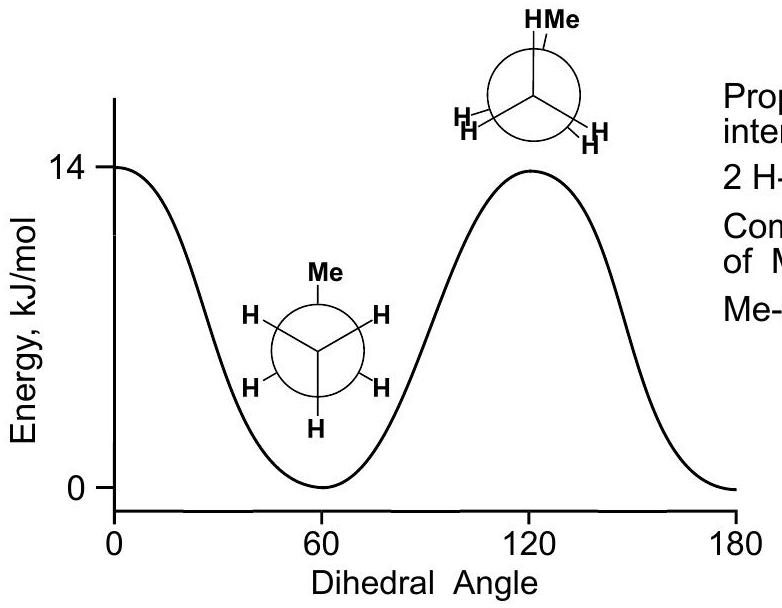
PROPANE
The energy plot is identical to ethane's, with a barrier of 14kJ//mol14 \mathrm{~kJ} / \mathrm{mol} instead of 12 .
Eclipsed
Staggered
Propane eclipsing interactions: 2H-H+1Me-H2 \mathrm{H}-\mathrm{H}+1 \mathrm{Me}-\mathrm{H}
Compute the value of Me-H eclipsing: Me-H=14-2**4\mathrm{Me}-\mathrm{H}=14-2 * 4 =6kJ//mol=6 \mathrm{~kJ} / \mathrm{mol}
How to think about rotations of two or more C-C bonds
(This section is not in McMurry, so it will not be on an exam or quiz.)
In McMurry and in this course, we use Newman projections to consider each rotation separately. In propane, butane, etc. an alkyl group attached to the C-C\mathrm{C}-\mathrm{C} bond is treated as a structureless lump that affects the C-C rotation barrier by steric interactions with nearby substituents.
Here is a more realistic treatment that illustrates how chemists think about molecules like propane. It will not be on an exam because it is not in McMurry, and you can safely skip the rest of this page.
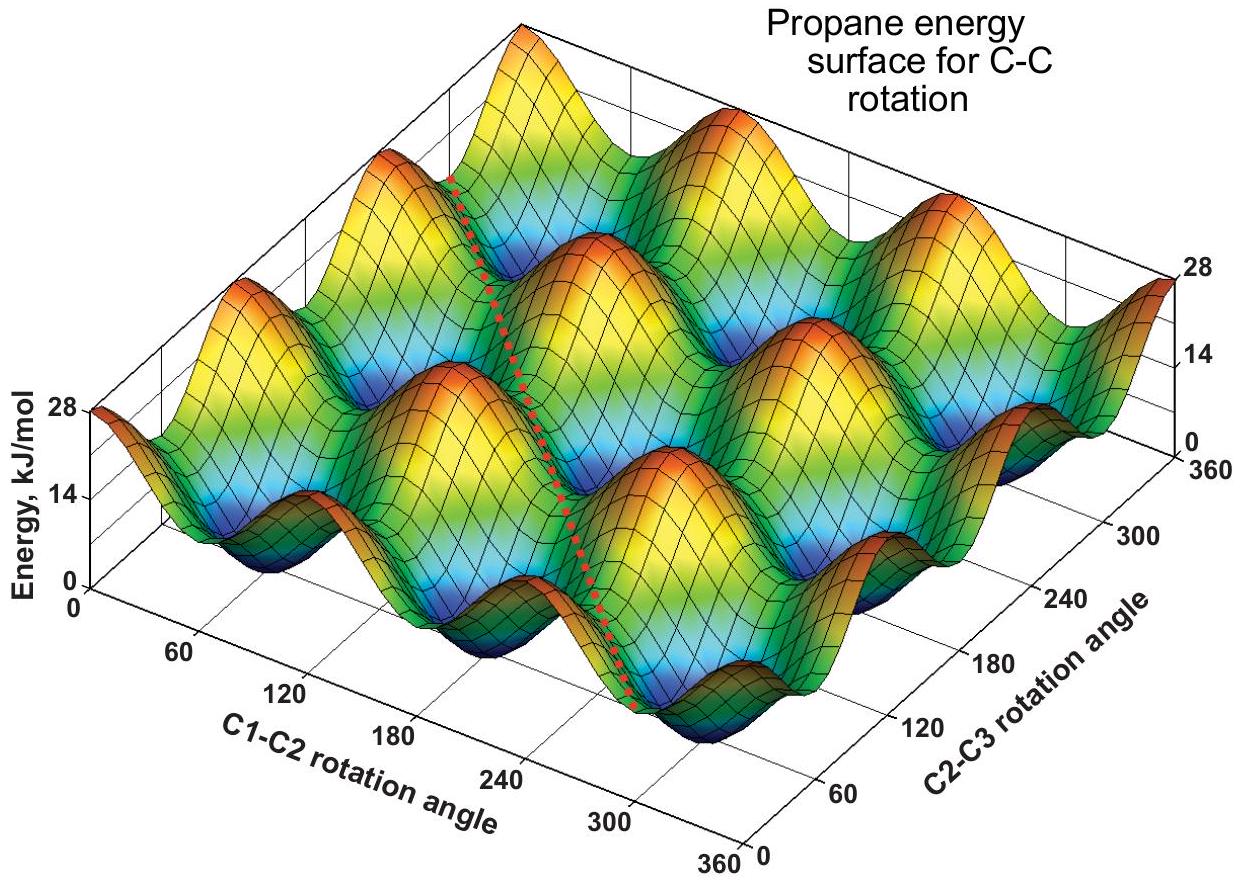
There are two C-C rotations, each with a barrier of 14kJ//mol14 \mathrm{~kJ} / \mathrm{mol}. Each rotation angle can have any value, and this leads to an energy surface, called an egg-carton surface. At the red peaks, the C_(1)C_(2)\mathrm{C}_{1} \mathrm{C}_{2} and C_(2)C_(3)\mathrm{C}_{2} \mathrm{C}_{3} rotations are both eclipsed, and at the blue minima they are both staggered. At other grid points one is staggered and the other eclipsed. The 1D curve above is recovered by picking any grid line and following it from 0 to 360^(@)360^{\circ} with the other angle constant. For example, the energy along the C_(1)C_(2)\mathrm{C}_{1} \mathrm{C}_{2} axis line ( C_(2)C_(3)=0\mathrm{C}_{2} \mathrm{C}_{3}=0 ) oscillates between 14 and 28 , and the energy along the adjacent grid line with C_(2)C_(3)=60\mathrm{C}_{2} \mathrm{C}_{3}=60 varies between 0 and 14 .
Molecular motion on an egg-carton surface
The diagonal red dotted line illustrates a path in which two CH_(3)\mathrm{CH}_{3} groups rotate together (cooperative motion). The path shown maintains a constant energy of 14kJ//mol14 \mathrm{~kJ} / \mathrm{mol} as the CH_(3)\mathrm{CH}_{3} groups rotate together with a constant 60^(@)60^{\circ} difference. This and similar paths imply that conformational change does not have to occur by hops between adjacent minima, but can also involve long-distance hops between nonadjacent minima. This example illustrates the conceptual advantage of energy surfaces over energy curves.
Egg-carton type energy surfaces (2D periodic surfaces) are useful for describing solid surfaces like metals, graphene, etc. They have been used in studying catalysis and material science.
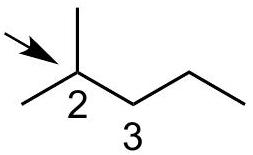
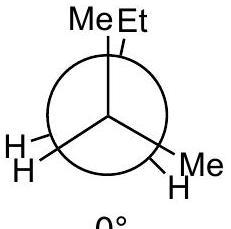
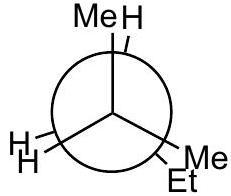
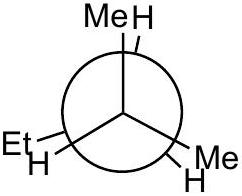
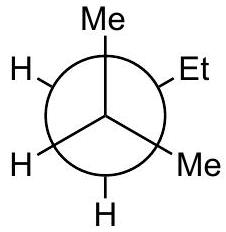
Practice problem:
Show a qualitative energy diagram for rotation about the C_(2)-C_(3)\mathrm{C}_{2}-\mathrm{C}_{3} bond of 2-methylpentane
Put C_(2)\mathrm{C}_{2} in front
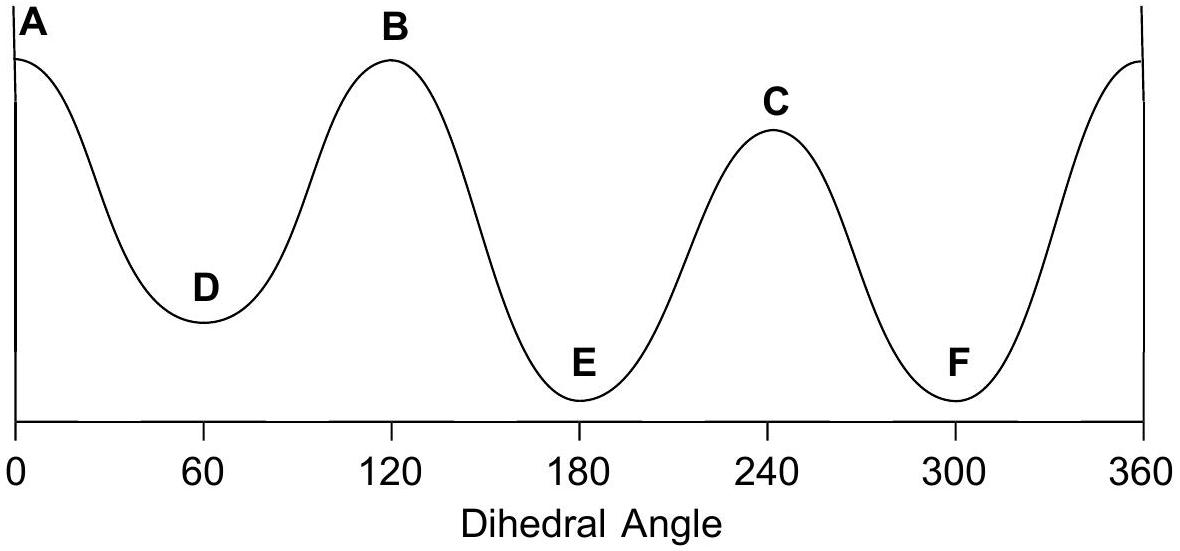
Draw 3 eclipsed and 3 staggered Newman projections, label them A - F, specify dihedral angles. 0^(@)0^{\circ} should be eclipsed.
Qualitatively assign highest and lowest energy eclipsed; same for staggered. Guiding principle: energy is lowest when the largest groups are separated.
Any eclipsed structure is higher in energy than any staggered.
Arrange A-FA-F on energy diagram so that the angle increases monotonically.